La absorción de radiación ultravioleta o visible por una especie atómica o molecular M se puede considerar como un proceso de dos etapas, la primera de ellas consiste en una excitación electrónica M + hν → M* El tiempo de vida de la especie excitada es breve (10 -8 a 10 -9s), su existencia se termina por alguno de los distintos procesos de relajación. El más común supone la conversión de la energía de excitación en calor M* → M + calor La relajación puede ocurrir también por la descomposición de M* para dar lugar a nuevas especies; dicho proceso se denomina reacción foto-química. En otros casos la relajación puede suponer la remisión de fluorescencia o fosforescencia. Es útil reconocer tres tipos de transiciones electrónicas y clasificar las especies absorbentes sobre esta base. Dichas transiciones incluyen (1) electrones σ, π y n, (2) electrones d y f, y (3) electrones de transferencia de carga. Las especies absorbentes que contienen electrones π, σ y n incluyen iones y moléculas orgánicas, así como algunos aniones inorgánicos. Todos los compuestos orgánicos son capaces de absorber radiación electromagnética porque todos contienen electrones de valencia que pueden ser excitados a niveles de energía superiores. Los enlaces sigma se pueden excitar solo con energía proveniente del ultravioleta al vacío. Las dificultades experimentales asociadas con el ultravioleta de vacío son significativas; como resultado de ello, la mayoría de las investigaciones espectrofotométricas de compuestos orgánicos se han realizado en la región de longitudes de onda superiores a 185 nm. La absorción de radiación visible y ultravioleta de longitud de onda larga está restringida a un número limitado de grupos funcionales (llamados cromóforos) que contienen electrones de valencia con energías de excitación relativamente bajas. Los espectros electrónicos de moléculas orgánicas que contienen cromóforos son, en general, complejos porque la superposición de las transiciones vibracionales sobre las transiciones electrónicas conduce a una compleja combinación de líneas solapadas; el resultado es una banda de absorción ancha que a menudo parece ser continua. Además de los electrones σ y π, muchas moléculas orgánicas contienen electrones no enlazantes. Estos electrones, que no participan en ningún enlace se designan con el símbolo n y están en gran parte localizados alrededor de átomos como oxígeno, halógenos, azufre y nitrógeno. Transiciones σ → σ*. En este caso, un electrón de un orbital σ enlazante de una molécula se excita al correspondiente orbital antienlazante mediante la absorción de radiación. Entonces se dice que la molécula se encuentra en el estado excitado σ*. Comparando con otras posibles transiciones, la energía requerida para que tenga lugar la transición σ → σ* es grande, correspondiendo a una frecuencia radiante de la región ultravioleta de vacío. Debido a las complicaciones experimentales asociadas a trabajar en el vacío el estudio de este tipo de absorción es muy limitado. Transiciones n → σ*. Los compuestos saturados que contienen pares de electrones no compartidos, n, son capaces de sufrir transiciones n → σ*. En general, estas transiciones requieren menos energía que las del tipo σ → σ* y se pueden producir por radiación de la región comprendida entre 150 y 250 nm, apareciendo, la mayoría de los picos de absorción, por debajo de 200 nm. Las absortividades molares asociadas a este tipo de absorción son de magnitud baja e intermedia y normalmente se encuentran entre 100 y 3000 L cm-1 mol -1 . Los máximos de absorción para las transiciones n → σ* tienden a desplazarse hacia longitudes de onda más cortas en presencia de disolventes polares como el agua o etanol. El número de grupos funcionales orgánicos con picos de este tipo en la región ultravioleta de fácil detección es relativamente pequeño. Transiciones n → π* y π → π*. La mayoría de las aplicaciones de espectroscopia de absorción en compuestos orgánicos se basan en transiciones de los electrones n y π al estado excitado π* porque la energía requerida para estos procesos produce picos de absorción dentro de una región espectral experimentalmente accesible (200 a 700nm). Ambas transiciones requieren la presencia de grupos funcionales no saturados que aportan los electrones π. Estrictamente hablando, es a estos centros absorbentes no saturados a los que se les aplica el término de cromóforos. Una diferencia entre ambos procesos es el efecto que tiene el disolvente en la longitud de onda de los picos. Los picos asociados con transiciones n → π* generalmente se desplazan hacia longitudes de onda más cortas (desplazamiento hipsocrómico o hacia el azul), cuando aumenta la polaridad del disolvente. Normalmente, aunque no siempre, se observa la tendencia opuesta en las transiciones π → π* (desplazamiento batocrómico o hacia el rojo). Los cromóforos orgánicos más comunes son: alqueno, alquino, carbonilo, carboxilo, amido, azo, nitro, nitroso y nitrato. La mayoría de los iones de los metales de transición absorben en la región ultravioleta o visible del espectro. Para la serie de lantánidos y actínidos, los procesos de absorción son el resultado de transiciones electrónicas de los electrones 4f y 5f; para los elementos de la primera y segunda serie de los metales de transición, los electrones responsables son los 3d y 4d.
Especies absorbentes que contienen electrones
La absorción de radiación ultravioleta y visible (200–800 nm) se restringe a un número limitado de grupos funcionales, llamados cromóforos, que contienen electrones de valencia con energías de excitación relativamente bajos.

Iones de los metales de transición
Serie de los lantánidos y actínidos
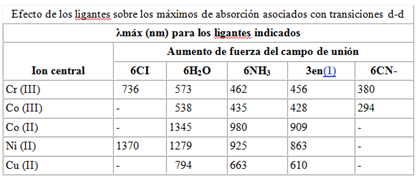
Los iones y complejos de los 18 elementos de las dos primeras series de transición son coloreados en uno de sus estados de oxidación o en todos ellos. Dependiendo las características colorimétricas del complejo; del tipo de ligando (agente complejante) y del estado de oxidación.
Para fines analíticos es el tipo más importante de absorción por especies inorgánicas, debido a que las absortividades molares de los picos son muy grandes (ξ < 105). Esta es una particular característica de los complejos inorgánicos denominados también, complejos de transferencia de carga.
Ejemplos:
Hierro III- ion tiocianato
Hierro II - (o-Fenantrolina)
Ion triyoduro I3-



